7 Antworten auf Fragen zur Impfstoffentwicklung
Die Entwicklung eines Impfstoffs, der eine gute Wirksamkeit und Verträglichkeit haben soll, ist ein zeit- und kostenintensiver Prozess. Das kann schon einmal bis zu 15 – 20 Jahre dauern und 300 – 800 Millionen Euro oder mehr kosten.1 Wir verraten 7 Fakten zur Impfstoffentwicklung!

Die notwendigen klinischen Studien bilden den wichtigsten Kostenfaktor im Entwicklungsprozess eines Impfstoffes:1
- Für eine Studie zur Sicherheit („extended safety“), für die etwa 10.000 – 60.000 Studienteilnehmer erforderlich sind, summieren sich die Kosten auf 50 – 150 Millionen Euro.1
- Für eine Studie zur Wirksamkeit („efficacy“) sind zumindest ein paar Tausend Studienteilnehmer nötig und es fallen Kosten von mindestens 50 Millionen Euro an.1
- Besondere Studien in Risikogruppen, wie z. B. chronisch Kranke oder Kinder, kosten mindestens 5 Millionen Euro.1
Wie lange es dauert, bis ein Impfstoff entwickelt ist, kann sehr unterschiedlich sein:
- Die frühere Entwicklung gängiger pädiatrischer Kombinationsimpfstoffe hat ca. 10 – 12 Jahre gedauert.1
- Die Entwicklung der Impfungen gegen humane Papillomviren (HPV) und Rotaviren hat jeweils etwa 15 Jahre gebraucht.1
- Die Entwicklung des Impfstoffs gegen Varizellen sowie die der Influenza-Lebendimpfung dauerte jeweils 25 – 30 Jahre.1
- Dank beschleunigter Zulassung ebenfalls relativ schnell verfügbar war ein Impfstoff gegen Ebola: Den auslösenden Startschuss gab eine große Ebola-Epidemie in Westafrika im Jahr 2014. Bereits im November 2019 wurde ein gentechnisch hergestellter, abgeschwächter Vektor-Lebendimpfstoff in Europa unter Vorbehalt zugelassen.2
- In Pandemiezeiten musste es schneller gehen: Die Impfstoffe gegen COVID-19 konnten dank neuer Plattform-Technologien3, früheren Erkenntnissen zu anderen Coronaviren und beschleunigten Abläufen und Verfahren4 binnen Monaten entwickelt und zugelassen werden3.
- Im Gegensatz dazu sind Impfstoffe gegen das Hepatitis-C-Virus (HCV) oder das humane Immundefizienz-Virus (HIV) bislang immer noch nicht verfügbar, obwohl seit über 30 Jahren daran geforscht wird.1
Warum ging die Entwicklung gängiger pädiatrischer Impfstoffe im Vergleich zu anderen Impfstoffen relativ schnell vonstatten? Der Grund: Es gab meist schon monovalente Präparate oder Ganzzell-Impfstoffe. Das Grundprinzip der Wirksamkeit des Impfstoffes stand hier aber schon länger fest. Sie wurden also nur als Kombinationsimpfstoffe oder Teilantigenimpfstoffe verbessert. Beispiele hierfür sind Toxoid-Impfstoffe gegen Tetanus und Diphtherie. Im Gegensatz dazu mussten für die Entwicklung „neuerer“ Impfstoffe, die z. B. gegen HPV und Rotaviren gerichtet sind, zuerst die schützenden Komponenten der Erreger identifiziert werden. Darüber hinaus kamen bei der Herstellung mit molekularbiologischen Technologien andere Methoden zum Einsatz als bei früheren Impfstoffen: Die meisten viralen Impfstoffe werden schon seit den 1960er Jahren in Zellkulturen hergestellt. Dazu gehören z. B. die Impfstoffe gegen Masern, Polio, Tollwut und Röteln.1
Ein weiterer Grund für die längere Entwicklungsdauer neuerer Impfstoffe: Die Anforderungen für die klinische Prüfung haben sich in den letzten Jahrzehnten – mit besonderem Fokus auf Impfstoffsicherheit – sehr verändert.1
Wie kam es dann aber zu der relativ schnellen Zulassung von Impfstoffen gegen z. B.COVID-19? Die Impfstoffe gegen COVID-19 durchliefen die gleichen Entwicklungs- und Zulassungsschritte wie andere Impfstoffe auch. Deutliche Optimierungen der Verfahrensabläufe haben jedoch zu einem Zeitgewinn bei der Entwicklung geführt. Dazu zählten z. B. eine frühe, kontinuierliche wissenschaftlich-regulatorische Beratung der Impfstoffentwicklung durch die Arzneimittelbehörden sowie ein Rolling-Review-Verfahren: Hier wurden schon während der klinischen Phase-III-Prüfung Datenpakete zur Vorab-Bewertung für die Zulassung vorgelegt. Zudem konnten die üblicherweise nacheinander ablaufenden klinischen Prüfungen kombiniert stattfinden.4
Sehenswert
Ein kurzes, informatives Video zum Thema Impfstoffentwicklung in der Corona-Pandemie des Bundesministeriums für Bildung und Forschung (BMBF) haben wir für Sie hier verlinkt.
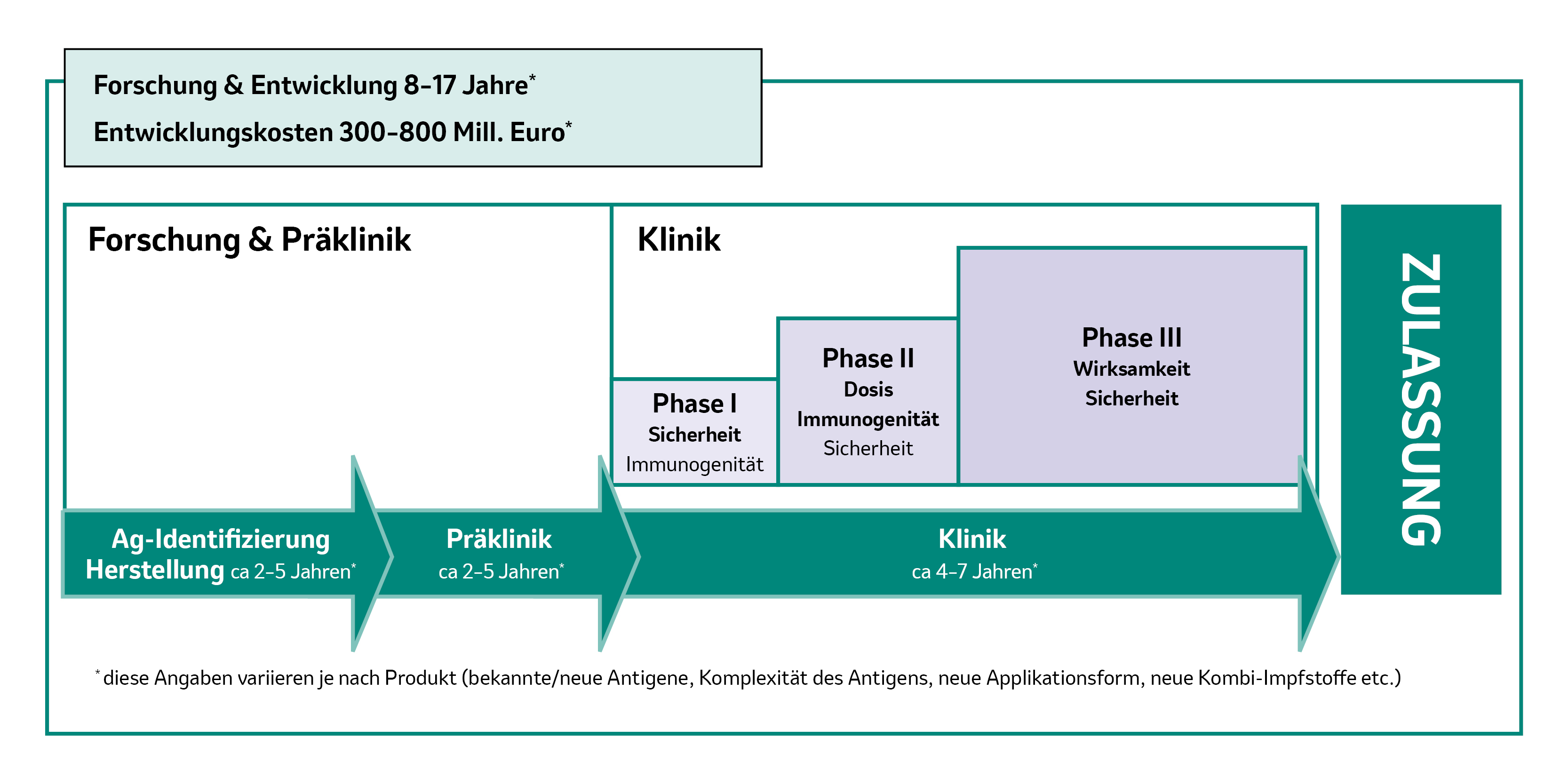
Abb. 1: Phasen der Impfstoffentwicklung – Dauer und Kosten. Abbildung modifiziert von MSD nach [1].
Es gibt eine ganze Reihe von wichtigen Faktoren, die bei der Frage, ob ein Impfstoff entwickelt wird oder eben nicht, eine entscheidende Rolle spielen:1
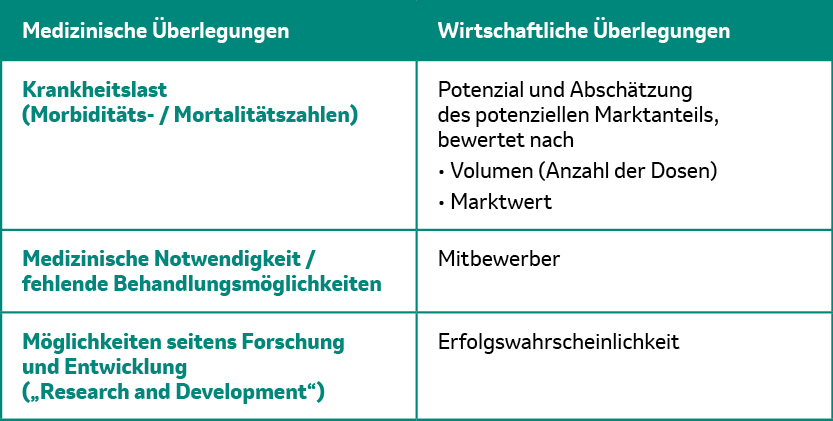
Tab. 1: Entscheidende medizinische und wirtschaftliche Faktoren für die Impfstoffentwicklung. Tabelle modifiziert von MSD nach [1].
Heutzutage wird systematisch wissenschaftlich an die Impfstoffentwicklung herangegangen. Dies umfasst die Identifikation und genaue Charakterisierung des Erregers bzw. der Erregerbestandteile, einschließlich einer Analyse zur Identifikation der jeweiligen spezifischen Antigene, die eine protektive Immunantwort auslösen können.1
Die Impfstoffherstellung selbst lässt sich unterschiedlich angehen. Zunächst muss entschieden werden, ob es ein Lebendimpfstoff oder ein inaktivierter („Tot“-)Impfstoff werden soll.1
Bei einem Lebendimpfstoff wird die gesamte Erregerstruktur, z. B. ein vollständiges, vermehrungsfähiges Virus als Impfantigen verwendet. Die Erreger werden dabei attenuiert, also in ihrer Virulenz so stark abgeschwächt, dass sie für einen gesunden Impfling keine Erkrankungsgefahr darstellen. Vorsicht ist hier jedoch bei immunsupprimierten Menschen geboten, da bei Lebendimpfstoffen von einem gesunden, funktionsfähigen Immunsystem ausgegangen wird.1
Bei Totimpfstoffen kann man mehrere Subtypen unterscheiden:1
- Beim Ganzzell-Impfstoff ist der Erreger als Gesamtheit erhalten, dabei jedoch inaktiviert.
- Beim Spalt-Impfstoff sind größere Teile eines zerstörten Erregers enthalten.
- Subunit-Impfstoffe enthalten nur noch bestimmte Antigene eines Erregers. Dazu gehören u. a. Toxoid-Impfstoffe z. B. gegen Tetanus und Diphtherie oder Polysaccharid-impfstoffe (Polysaccharide aus den Bakterienkapseln, etwa von Pneumokokken).
Bei den Vektor-Impfstoffen dient ein abgeschwächtes (attenuiertes) Virus als Transportmittel (Vektor) für einen ungefährlichen Teil der Erbinformation eines Erregers in wenige Körperzellen.3 Das Vektor-Virus kann dabei vermehrungsfähig oder auch nicht vermehrungsfähig sein und überträgt den Bauplan für ein oder auch mehrere Antigene.5 Als Vektor-Viren können z. B. bestimmte Adenoviren eingesetzt werden.6 Diese Vektorimpfstoffe können keine Erkrankung beim Menschen auslösen.7
mRNA-Impfstoffe enthalten Teile der Erbinformation eines Virus in Form einer Boten-RNA (messenger-RNA; mRNA). Um die Aufnahme der mRNA in die Zellen möglich zu machen, wird sie mit Lipidstoffen, also kleinen Fetttröpfchen umhüllt. Es entstehen sogenannter mRNA-Lipidnanopartikel. Nach der Injektion in den Muskel verschmelzen diese mit der Zellmembran der Muskelzellen, wodurch die mRNA in die Zellen gelangt. Die Nanopartikel sind nicht zellschädigend und stellen keine Gefahr für den Körper dar. In den Zellen wird die mRNA als Vorlage genutzt, um die darin codierten Virusproteine zu produzieren. Hierbei wird die mRNA nicht in DNA (Desoxyribonukleinsäure) umgebaut, sie hat somit keinen Einfluss auf die menschliche DNA. Anschließend werden die fertigen Proteine dem Immunsystem präsentiert. Dieses reagiert mit einer gezielten Antikörperbildung gegen das Virus-Protein (humorale Immunantwort) und einer zellulären Abwehr gegen mit dem jeweiligen Virus infizierte Zellen (zelluläre Immunantwort).8
Darüber hinaus gibt es mehrere Methoden zur Induktion oder Verbesserung einer protektiven Immunantwort nach Impfung. Dazu zählen z. B. die Konjugation von Antigenen an Trägerproteine, die Verwendung von immunstimulierenden Adjuvantien, die Verwendung von „Virus-Like Particles“ (VLP) sowie die reverse Vakzinologie zur Identifikation von potenziellen Impfstoffantigenen.1
Für Impfstoffe gelten genau wir für neue Arzneimittel – unabhängig vom Wirkstoff – immer standardisierte Entwicklungsgrundsätze. Diese gelten somit auch für Impfstoffe. Gibt es am Anfang im Labor noch 10.000 Kandidatensubstanzen, dann bleiben aufgrund der hohen Qualitätsanforderungen vielleicht noch 250 davon übrig, die in die präklinische Phase gehen. Am Ende bleiben vielleicht 5, die in die klinische Entwicklung gehen – und auch hier bedeutet das keine automatische Zulassung.1
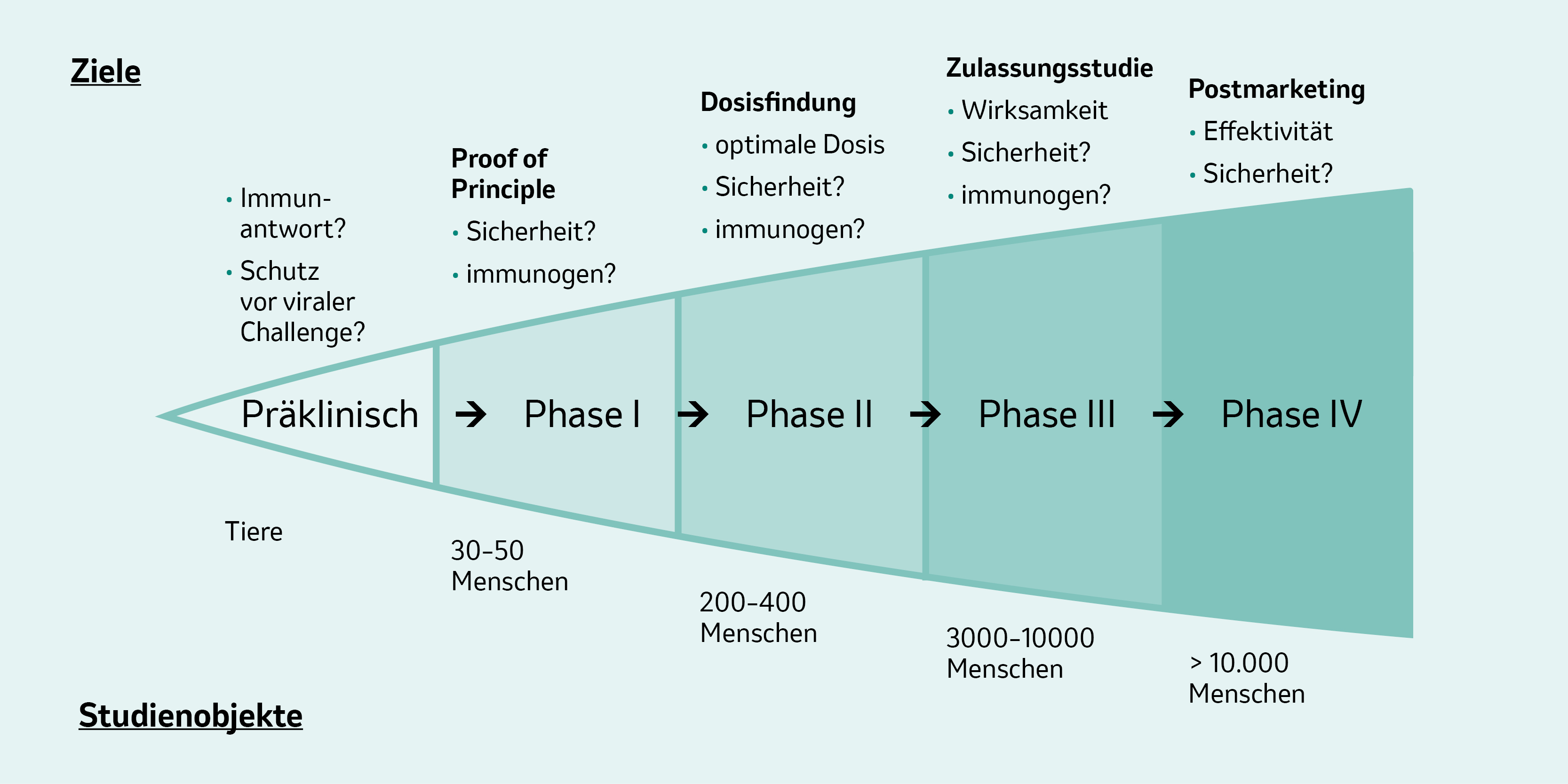
Abb. 2: Schematischer Ablauf der Phasen und Zahl der benötigten Studienteilnehmer bei der Impfstoffentwicklung. Abbildung modifiziert von MSD nach [1].
Präklinische Phase
Vor Beginn einer klinischen Studie, werden erste Information zur Wirksamkeit, Immunogenität und Sicherheit des verwendeten Antigens tierexperimentell erhoben. Hier geht es vor allem um die Dosis, die Impfstoffzusammensetzung, ggf. um Zusatz von Adjuvantien/Immunmodulatoren und die optimale Applikationsroute (i.d., s.c., i.m.). Allerdings sind die Etablierung von Tiermodellen und generell auch die Übertragung präklinischer Ergebnisse auf den Menschen nicht immer einfach und auch nicht 1:1 möglich.1
Klinische Phase I
In Phase-I-Studien werden Impfstoffe erstmals am Menschen („first in human“) geprüft: Man tastet sich anhand einer geeigneten Startdosis und einer relativ kleinen Probandenzahl (z. B. 30) an die Eigenschaften des Impfstoffkandidaten heran. Das primäre Ziel ist dabei Erkenntnisse zur Sicherheit und Verträglichkeit zu erhalten.1
Zusätzlich:
- werden vorläufige Daten zur Immunogenität erhoben.1
- wird die gewählte Dosis und Applikationsart evaluiert und ggf. adaptiert.1
- werden bei lebend-attenuierten Impfstoffen ggf. auch erste Informationen zu Erregerausscheidung, Transmission und genetischer Stabilität erhoben.1
- wird grundsätzlich eine Basis geschaffen, um nachfolgende definitivere und größere Studien optimal planen zu können.1
Bei der Erstanwendung am Menschen muss die Impfdosis (Antigenmenge bzw. die Verwendung von Adjuvantien) so gewählt werden, dass sie verträglich genug ist (lokal und systemisch), um eine Gefährdung möglichst zu vermeiden, aber dennoch immunogen genug, um die erwünschte Immunantwort nachweisbar auszulösen.1
So werden in Phase I vorwiegend sehr häufige Nebenwirkungen erfasst.1
Klinische Phase II
Phase-II-Studien haben meist mehrere 100 Studienteilnehmer aus der Zielpopulation. Ziel dieser Studien ist es, die Immunogenität und Sicherheit der Testvakzine zu bestätigen. Dabei geht es besonders um die Dosis-, Applikations- und Impfschema-Optimierung für große Phase-III-Studien. Zudem wird der Einfluss von unterschiedlichen Variablen auf die Immunantwort untersucht, z. B. werden Alter, Geschlecht, präexistierende Antikörper und der Prozentsatz der Responder erhoben.1
Phase-I- und Phase-II-Studie dauern jeweils etwa 1 – 1 ½ Jahre und können getrennt oder als kombinierte Phase-I/II-Studien durchgeführt werden.1
Klinische Phase III
Die klinische Phase III kann bis zu 4 Jahre dauern. In dieser Phase nehmen bereits einige Tausend freiwillige Teilnehmer an den Impfstudien teil. Phase-III-Studien werden zum Teil auch als „Zulassungsstudien“ bezeichnet. Dabei geht es um den Nachweis eines positiven Nutzen-Risiko-Profils – also um den bestmöglichen Schutz bei vertretbarem Risikoprofil.1
Phase-III-Studien dienen der Untersuchung von Sicherheit und Wirksamkeit eines Impfstoffs für die jeweilige Indikation und Zielpopulation. Außerdem wird durch sogenannte „lot to lot consistency“-Studien die Konsistenz in der Herstellung von Charge zu Charge klinisch überprüft.1
Klinische Phase IV
Die klinische Phase IV umfasst sogenannten Postmarketing-Studien für den bereits zugelassenen Impfstoff. Für gewöhnlich werden darin mehr als 10.000 Teilnehmer eingeschlossen. Hierbei handelt es sich z. B. um Studien zum Sicherheitsprofil, denn: Seltenere Nebenwirkungen können auch mit mehreren Tausend Teilnehmern in Phase-III-Studien nicht sicher erfasst werden.1
Diese Studien können – im Zuge der Pharmakovigilanz – u. a. als nicht-interventionelle Studien oder als Fall-Kontroll-Studien durchgeführt werden. Dabei wird das Sicherheitsprofil nach Zulassung des Impfstoffs ständig durch den Zulassungsinhaber und die zuständige Behörde überwacht.1
Das behördliche Zulassungsverfahren überprüft anhand der regulatorischen Angaben:1
- Daten zur Herstellung
- Präklinische Daten
- Klinische Daten
- Informationen zur Pharmakovigilanz
- Informationen zu den Impfstoffantigenen
- Informationen zu allen anderen Inhaltsstoffen
Ein Arzneimittel wird zugelassen, wenn es ein positives Nutzen-Risiko-Verhältnis zeigt.1
Impfstoffe gehören dabei zu den Chargenfreigabe-pflichtigen Arzneimitteln: Jede Charge muss von einem staatlichen Arzneimittelkontrolllabor („Official Medicines Control Laboratory“ – OMCL) eines EU- bzw. EWR- (Europäischer Wirtschaftsraum-)Landes geprüft und freigegeben werden.1
Solang ein Produkt auf dem Markt ist, werden auch laufend Nebenwirkungsmeldungen evaluiert. Dazu ist es für den Zulassungsinhaber verpflichtend, der Behörde regelmäßige Sicherheitsberichte – sogenannte „Periodic Safety Update Reports“ (PSUR) – vorzulegen.1
Eventuell neu auftretende Risiken müssen anlassbezogen und umgehend evaluiert werden. Zudem müssen Risiko-minimierende Maßnahmen gesetzt werden. Das können beispielsweise eine Änderung der Produktinformation oder Informationsschreiben an Ärzte sein.1
Lesenswert
Welche Herstellungsschritte sind für die Impfstoffentwicklung erforderlich? An welchen Stellen erfolgen Qualitätskontrollen? Antworten gibt Ihnen unsere anschauliche Infografik Impfstoffherstellung: Der komplexe Weg eines Impfstoffs!
Mit welchen stichhaltigen Argumenten kann man versuchen, die Bedenken impfkritischer Patient:innen abzubauen? Wir geben Ihnen mögliche Entgegnungen auf 10 häufige Impf-Einwände für den Umgang mit impfkritischen Patienten – Teil 2 an die Hand.
Quellen
- Wiedermann U., et al. Entwicklung von Impfstoffen. Österreichische Ärztezeitung Nr. 23 – 24. 15.Dezember 2017.
- Paul-Ehrlich-Institut (PEI). Weltweit erster Ebola-Impfstoff zugelassen. Online verfügbar unter: https://www.pei.de/DE/newsroom/hp-meldungen/2019/191113-erster-impfstoff-schutz-vor-ebola-zulassung-in-eu.html [eingesehen am 17.09.2025].
- Bundesinstitut für Öffentliche Gesundheit (BIÖG). Das Impfbuch für alle. Online verfügbar unter: https://shop.bioeg.de/das-impfbuch-fuer-alle/ [eingesehen am 17.09.2025].
- Paul-Ehrlich-Institut (PEI). FAQ – Häufig gestellte Fragen: Coronavirus. Online verfügbar unter: https://www.pei.de/DE/service/faq/weitere/faq-coronavirus-node.html [eingesehen am 17.09.2025].
- Bundesinstitut für Öffentliche Gesundheit (BIÖG). Welche unterschiedlichen Impfstoffe gibt es? Stand: 21.08.2024. https://www.infektionsschutz.de/impfen/wissenswertes-zum-impfen/impfstoffe/ [eingesehen am 17.09.2025].
- Hildt E. Plattformtechnologien in der Impfstoffentwicklung. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2025;68(4):368-377. DOI:10.1007/s00103-025-04024-6.
- Bundesministerium für Gesundheit (BMG). Ratgeber Impfen. Stand: März 2025. https://www.bundesgesundheitsministerium.de/service/publikationen/details/ratgeber-impfen.html [eingesehen am 17.09.2025].
- Robert Koch-Institut (RKI). Impfstofftypen. Stand: 15.04.2025. https://www.rki.de/SharedDocs/FAQs/DE/Impfen/COVID-19/FAQ_Liste_Impfstofftypen.html [eingesehen am 17.09.2025].
DE-NON-02539 10/25